
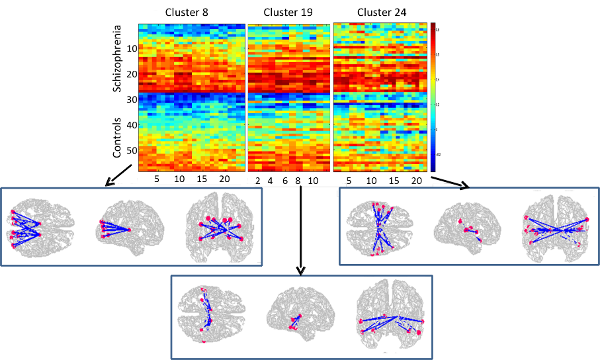
Brain connectivity in schizophrenia
Brain connectivity differences between schizophrenia and healthy subjects are typically studied by testing one edge at a time. The noise inherent in fMRI data and the inability to leverage network structure has resulted in largely inconsistent findings. In this project we developed a systematic approach by leveraging the underlying network structure and found that this helps in handling noise and results in consistent findings. The results are reported in a recent Human Brain Mapping paper.
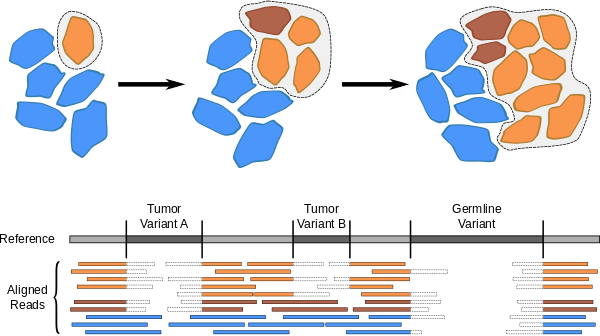
Genetic heterogeneity in tumors
Genomic instability and extreme proliferation of cancer cells can lead to a high degree of genetic heterogeneity within tumor tissue. Next-generation sequencing data from tumor samples will usually contain mixtures of DNA from diverse tumor cell populations, as well as from normal somatic cells, each with their own set of variants. We developed software that predicts these heterogeneous variants, along with estimating their respective frequencies. For more information, see our recent BMC Genomics paper.
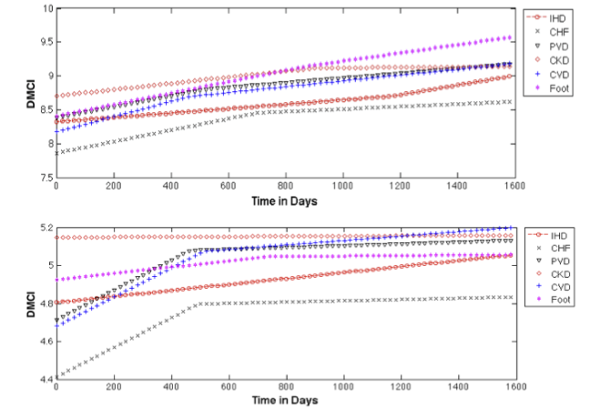
Mining electronic health records
With the implementation of electronic health records (EHRs), data are available to investigate actual use and effectiveness over time within and across subpopulations. As a first step, we developed a diabetes risk score (DRS) using EHR data and modeled trajectories for development of diabetic complications based on groups of patients with varying diabetic problems at baseline. Results presented at AMIA 2014.
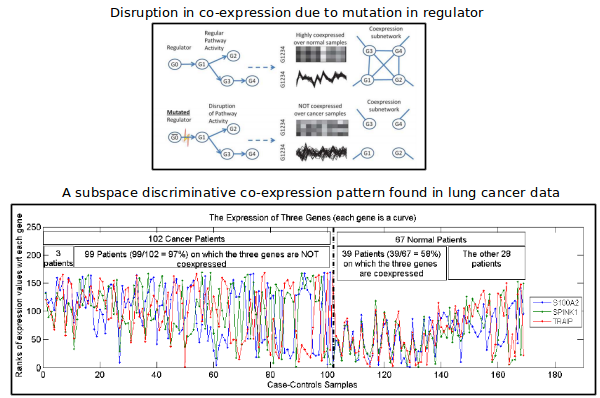
Differential coexpression of genes
A differential coexpression pattern consists of a set of genes that have substantially different levels of coherence of their expression profiles across the two sample-classes. Biologically, a differential coexpression patterns may indicate the disruption of a regulatory mechanism possibly caused by disregulation of pathways or mutations of transcription factors. We extended differential coexpression analysis by defining a subspace differential coexpression pattern. The method and results are reported in our PSB 2010 paper.
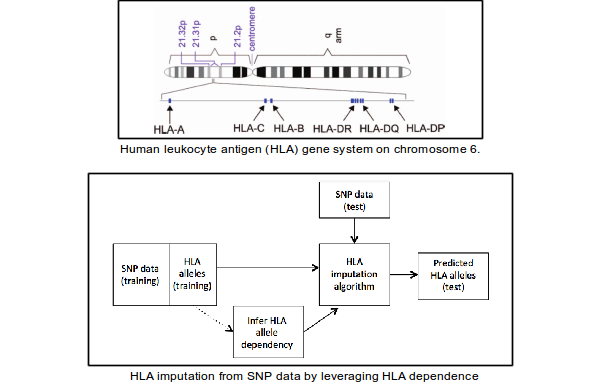
Genotyping HLA
The Human Leukocyte Antigen (HLA) gene system is the most polymorphic region of the human genome, containing some of the strongest associations with autoimmune, infectious, and inflammatory diseases. It plays a crucial role in hematopoietic stem cell transplantation, where patients and donors are matched with respect to their HLA genes to maximize the chances of a successful transplant. The availability of HLA data is, therefore, of high importance to clinicians and researchers. However, due to its high polymorphism, obtaining it is time- and cost-prohibitive. Our approach for the prediction of HLA genes from widely available Single Nucleotide Polymorphism (SNP) data is presented in our BCB'13 paper.
About Us
We are a research group in the Department of Computer Science & Engineering at the University of Minnesota that specializes in developing novel data mining and machine learning techniques to analyze a wide variety of biomedical data sets. For more information, see our Research page.