Subspace Differential Coexpression Analysis: Problem Definitions and
A General Approach
Gang Fang,
Rui Kuang,
Gaurav Pandey,
Michael Steinbach,
Chad L. Myers and
Vipin Kumar
Proceedings of the 15th
Pacific Symposium
on Biocomputing, 15:145-156, Jan 2010. (paper, slides)
Also highlighted by
RECOMB Satellite Meeting on Systems Biology for oral
presentation (video (TBA))
Correspondence: Gang Fang (gangfang cs umn edu)
Last updated:
01/18/2010
Source codes, sample datasets and detailed
instructions : [sdc_0p21.zip (6.87MB)]
Pre-processing:
Conversion from
standard microarray data files to the input format required by the SDC mining
algorithm
Mining algorithm: (currently only linux version for now)
An
Apriori-based SDC pattern mining algorithm: because
of the antimonotonicity of Refined-SDC, given a threshold d, this Apriori-based
framework guarantees to discover ALL and ONLY the patterns with RSDC >= d,
i.e. with guaranteed power of subspace differential coexpression.
Post-processing:
Visualization of SDC patterns as used in the paper
Summarization from a set
of patterns to a less redundant set
Conversion of SDC
patterns into Cytoscape network format
for global visualization of SDC patterns.
Other files and information:
Detailed data information:
The RMA normalized and Combined Gene Expression Data can be downloaded from here:
Expression value version (a 169 by 8787 matrix,
8MB), Rank-converted version (a 169 by 8787
matrix, 5MB), Class labels (0: cancer class, 1:
normal class) and Gene Symbols. Links to the original formats of the three lung
cancer datasets: (Stearman
et al. 2005,
Su
et al. 2007, Bhattacharjee
et al. 2001)
Cancer-gene list:
The 2622 known cancer-related genes can be
downloaded from here: CancerGeneList2622.
(This is the union of the two lists respectively downloaded from
http://cbio.mskcc.org/cancergenes/Select.action on October 2008 and June
2009)
Abstract
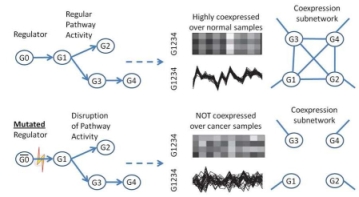
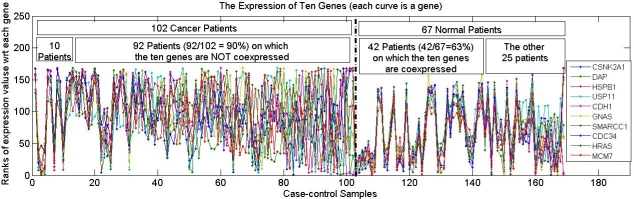
In this paper, we study methods to identify differential coexpression patterns in case-control gene expression data. A differential coexpression pattern consists of a set of genes that have substantially different levels of coherence of their expression profiles across the two sample-classes, i.e., highly coherent in one class, but not in the other. Biologically, a differential coexpression patterns may indicate the disruption of a regulatory mechanism possibly caused by disregulation of pathways or mutations of transcription factors. A common feature of all the existing approaches for differential coexpression analysis is that the coexpression of a set of genes is measured on all the samples in each of the two classes, i.e., over the full-space of samples. Hence, these approaches may miss patterns that only cover a subset of samples in each class, i.e., subspace patterns, due to the heterogeneity of the subject population and disease causes. In this paper, we extend differential coexpression analysis by defining a subspace differential coexpression pattern, i.e., a set of genes that are coexpressed in a relatively large percent of samples in one class, but in a much smaller percent of samples in the other class. We propose a general approach based upon association analysis framework that allows exhaustive yet efficient discovery of subspace differential coexpression patterns. This approach can be used to adapt a family of biclustering algorithms to obtain their corresponding differential versions that can directly discover differential coexpression patterns. Using a recently developed biclustering algorithm as illustration, we perform experiments on cancer datasets which demonstrates the existence of subspace differential coexpression patterns. Permutation tests demonstrate the statistical significance for a large number of discovered subspace patterns, many of which can not be discovered if they are measured over all the samples in each of the classes. Interestingly, in our experiments, some discovered subspace patterns have significant overlap with known cancer pathways, and some are enriched with the target gene sets of cancer-related microRNA and transcription factors. The source codes and datasets used in this paper are available at http://vk.cs.umn.edu/SDC/ .
The quantitative enrichment results are here.
In the paper, we discussed the patterns discovered on the rank-converted data, while the patterns discovered on expression value data is presented here.
Parameters details are provided in the
paper.
If any questions, please email : Gang Fang, gangfang cs umn edu